



Until recently, immune mechanisms in the pathogenesis of Alzheimer’s disease have been largely overlooked. Following the pioneer work of McGeer, Rogers and Griffins it is today generally accepted that cellular and molecular components of immune system reactions are associated with AD (e.g. McGeer and Rogers, 1992, Griffins, 1989).
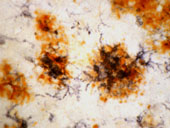
Activated microglia (the brain's representatives of the phagocytic cells that are designed to clean up debris and foreign bacteria) surround senile plaques and extracellular neurofibrillary tangles. AD lesions are characterized by the presence of a series of inflammatory mediators, including cytokines, chemokines, proteases, adhesion molecules, free radicals, pentraxins, prostaglandins, anaphylatoxin, and activated complement proteins (McGeer et al., 1995; McGeer and McGeer, 2002). (Figure: On double immunostained section, dark blue HLA-DR immunoreactive microglia and orange-brown immunostained beta amyloid are visible in senile plaques. From the laboratory of Pat McGeer).
Using specific immunohistochemical markers, both T-helper/inducer and T-cytotoxic/suppressor lymphocytes have been observed. Of particular importance is the association of the membrane attack complex (MAC, C5b-9) intended to lyse foreign cells, such as bacteria with dystrophic neurites (McGeer and Rogers, 1992; Webster et al., 1997). The conclusion that inflammation exacerbates AD pathology is now supported by more than 20 epidemiological studies showing that individuals are spared AD if they have been taking anti-inflammatory drugs or have suffered from unrelated conditions for which such drugs are routinely used (McGeer et al., 1996, Veld et al., 2000). This effect has been particularly evident in people using nonsteroidal anti-inflammatory drugs (NSAIDs), which are widely used agents in various arthritic conditions. Three large epidemiological studies showed a reduction of risk of 55-80% for AD (Stewart et al., 1997; Zandi et al., 2000, Veld et al., 2000).